Die Chorea-Akanthozytose ist eine sehr seltene angeborene Erkrankung mit den Hauptmerkmalen Akanthozytose mit neurologischen Auffälligkeiten und normalen Lipoproteinen im Blutserum. Sie gehört zu den Neuroakanthozytosen.
Synonyme sind: ChAc; Choreoakanthozytose; Levine-Critchley-Syndrom
Die Namensbezeichnung wurde im Jahre 1985 von Tetsuo Sakai und Mitarbeiter vorgeschlagen und bezieht sich auf den Autoren der Entdeckung des Krankheitsbildes im Jahre 1960 durch den US-amerikanischen Arzt Irvine M. Levine und dessen Erstbeschreibung aus dem Jahre 1964 und 1968 sowie auf den britischen Neurologen MacDonald Critchley.
Verbreitung
Die Häufigkeit ist nicht bekannt, es sind wohl weltweit etwa 500–1000 Betroffene, gehäuft in Japan bekannt. Die Vererbung erfolgt autosomal-rezessiv oder autosomal-dominant.
Ursache
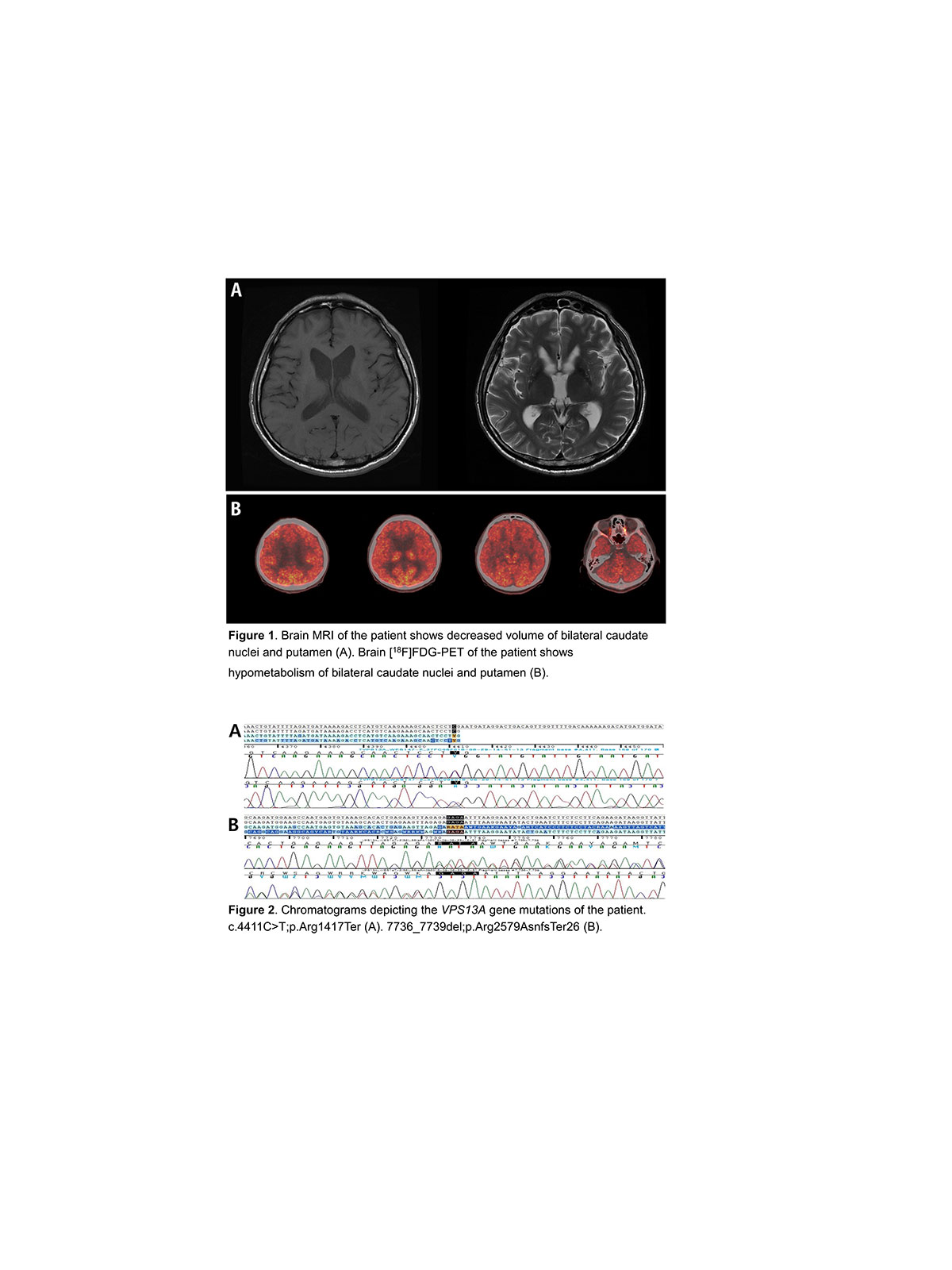
Der Erkrankung liegen verschiedene Mutationen im VPS13A-Gen auf Chromosom 9 Genort q21.2 zugrunde, welches für Chorein kodiert.
Klinische Erscheinungen
Klinische Kriterien sind:
- Manifestation im Erwachsenenalter, bei etwa einem Drittel Krampfanfälle als Erstzeichen
- Akanthozytose
- neurologische Auffälligkeiten meist im Erwachsenenalter wie Tourette-Syndrom, Chorea Huntington, Dyskinesie im Mund- und Gesichtsbereich, oft Zungen- und Lippenbisse, Grimassen, Dysarthrie
- Hyperkinesen der Gliedmaßen
- mitunter parkinsonartig
- oft distale Muskelatrophie mit abgeschwächten Eigenreflexen (MER)
- schwankendes Gangbild
- auch Sensibilitätsstörungen
- Creatin-Kinase im Serum meist erhöht
- selten Blasenfunktionsstörung
- Liquor cerebrospinalis und Serumlipoproteine normal
- Nervenleitgeschwindigkeit normal
Diagnose
Die Diagnose kann schwierig sein. Wichtige Hinweise sind selbstverletzendes Verhalten. Die Akanthozytose muss nicht immer nachweisbar sein, die Western-Blot-Analyse ergibt das Fehlen von Chorein in den Erythrozyten.
Die Magnetresonanztomographie ergibt eine Atrophie des Striatum, insbesondere des Kopfes des Nucleus caudatus mit vermindertem Glukosestoffwechsel.
Differentialdiagnose
Abzugrenzen sind:
- Bassen-Kornzweig-Syndrom (AR)
- McLeod-Syndrom (XR)
- Chorea Huntington
- Chorea-Huntington-ähnliche Krankheiten
- juvenile Form der Parkinson-Krankheit
- Tourette-Syndrom
Therapie
Bislang ist lediglich eine symptombezogene Behandlung möglich. Tiefe Hirnstimulation scheint eine Verbesserung zu bringen.
Verlauf
Die Aussichten sind wegen des fortschreitenden Verlaufes ungünstig. Krampfanfälle oder eine vegetative Fehlfunktion kann zum plötzlichen Ableben führen.
Literatur
- B. Bader, C. Dobson-Stone, A. Velayos-Baeza, A. Monaco, R. Walker, A. Danek: Chorea-Akanthozytose: Genetik und Verlauf von 106 Patienten. In: Aktuelle Neurologie. 35, 2008, doi:10.1055/s-0028-1086535.
- L. P. Hiersemenzel, S. Johannes, P. Themann, B. Hofferberth: Die Choreoakanthozytose. Ein neurologisch-hämatologisches Syndrom. In: Der Nervenarzt. Band 67, Nummer 6, Juni 1996, S. 490–495. PMID 8767204 (Review).
Weblinks
- Medline Plus
- Rare Diseases
- Gene Reviews
Einzelnachweise